Visium Human DLPFC Analysis
Source:vignettes/illustration_on_visium_data.Rmd
illustration_on_visium_data.RmdApplication of Spacelink to Visium Human DLPFC Dataset
Here, we demonstrate how to perform SVG analysis using the
spacelink package with a Visium human dorsolateral
prefrontal cortex (DLPFC) dataset. The dataset is available here, and we use
sample 151673. Cell-type proportions were estimated using RCTD with the CosMx
Human Frontal Cortex dataset as a reference. The processed example
data, provided as Visium_human_DLPFC,
includes gene expression, spot coordinates, and the estimated cell type
proportions.
We walk through the full Spacelink workflow in two stages: (1) global tissue-level SVG detection, which tests for spatial variability across all spots regardless of cell type, and (2) cell-type-specific SVG detection, which tests for spatial variability within a specific cell population.
2. Load Example Dataset
Here we load the built-in example dataset for Visium Human DLPFC (sample 151673). It comes with three pieces of information we’ll use throughout the analysis:
-
counts: Gene expression matrix (33,538 genes × 3,639 spots) -
spatial_coords: Spatial coordinates for each spot -
cell_type_proportions: Estimated cell type compositions via RCTD (9 cell types)
data(Visium_human_DLPFC)
counts <- Visium_human_DLPFC$counts
spatial_coords <- Visium_human_DLPFC$spatial_coords
cell_type_proportions <- Visium_human_DLPFC$cell_type_proportions
head(counts[,1:10])
## 6 x 10 sparse Matrix of class "dgTMatrix"
## [[ suppressing 10 column names ‘AAACAAGTATCTCCCA-1’, ‘AAACAATCTACTAGCA-1’, ‘AAACACCAATAACTGC-1’ ... ]]
## MIR1302-2HG . . . . . . . . . .
## FAM138A . . . . . . . . . .
## OR4F5 . . . . . . . . . .
## AL627309.1 . . . . . . . . . .
## AL627309.3 . . . . . . . . . .
## AL627309.2 . . . . . . . . . .
head(spatial_coords)
## pxl_col_in_fullres pxl_row_in_fullres
## AAACAAGTATCTCCCA-1 9791 8468
## AAACAATCTACTAGCA-1 5769 2807
## AAACACCAATAACTGC-1 4068 9505
## AAACAGAGCGACTCCT-1 9271 4151
## AAACAGCTTTCAGAAG-1 3393 7583
## AAACAGGGTCTATATT-1 3665 8064
head(cell_type_proportions)
## endothelial L2_3 L4 L6 oligodendrocyte OPC astro Inh microglia
## AAACAAGTATCTCCCA-1 2.331552e-02 0.86383709 9.941496e-05 8.994572e-05 0.008841064 8.994572e-05 0.037094727 0.060440671 0.0061916188
## AAACAATCTACTAGCA-1 7.952809e-02 0.55623941 1.284478e-04 1.413909e-02 0.001048564 1.284478e-04 0.083634323 0.264896732 0.0002568957
## AAACACCAATAACTGC-1 4.144100e-05 0.07909515 4.144100e-05 8.593492e-02 0.826629114 7.799136e-05 0.006276291 0.000124323 0.0017793364
## AAACAGAGCGACTCCT-1 4.419690e-04 0.70660998 1.437067e-04 1.437067e-04 0.012711896 1.437067e-04 0.119882025 0.133047245 0.0268757663
## AAACAGCTTTCAGAAG-1 1.398113e-02 0.52170271 8.882893e-05 2.874930e-01 0.050319200 8.882893e-05 0.026019353 0.096453571 0.0038533732
## AAACAGGGTCTATATT-1 8.872097e-05 0.37248441 8.872097e-05 3.500929e-01 0.206785555 8.872097e-05 0.029330741 0.040862758 0.00017744193. Preprocessing
Before running the analysis, we do a bit of cleaning. First, we remove mitochondrial genes since high mitochondrial expression usually signals low-quality or damaged cells rather than real biology. We also drop genes that are expressed in very few spots (less than 0.5%), as they don’t carry enough information to be useful. Finally, we normalize the counts using SCTransform, which corrects for differences in sequencing depth across spots.
counts <- counts[!grepl("(^MT-)|(^mt-)", rownames(counts)),]
counts <- counts[apply(counts >= 3, 1, sum) >= ncol(counts)*0.005,]
dim(counts)
## [1] 3309 3639
seurat_obj <- CreateSeuratObject(counts = counts)
seurat_norm = SCTransform(seurat_obj, vst.flavor = "v2", verbose = FALSE)
normalized_counts <- seurat_norm@assays$SCT$data
head(normalized_counts[,1:10])
## 6 x 10 sparse Matrix of class "dgCMatrix"
## [[ suppressing 10 column names ‘AAACAAGTATCTCCCA-1’, ‘AAACAATCTACTAGCA-1’, ‘AAACACCAATAACTGC-1’ ... ]] ##
## HES4 0.6931472 . . . . . . . . .
## ISG15 . . . . 0.6931472 . . 0.6931472 . .
## AGRN 0.6931472 1.386294 . . 0.6931472 . . 0.6931472 . .
## SDF4 0.6931472 . . 0.6931472 . . . . 0.6931472 .
## ACAP3 0.6931472 . . . . . . . 0.6931472 .
## DVL1 . . . . . 0.6931472 . . . .4. Run Global Spacelink
Now we’re ready to run the main analysis. Here we use spacelink
to identify spatially variable genes (SVGs) across the tissue. We’ll
test this on 6 example genes to keep things simple. The two numbers to
pay attention to in the results are padj (whether the
spatial autocorrelation is statistically significant) and
ESV_adj (how strong that spatial autocorrelation is, on a
scale from 0 to 1). Note that the padj and
ESV_adj values will differ when running the analysis on the
full gene set, because the Benjamini–Hochberg adjustment is applied
across all genes. In this example, the adjustment is performed using
only the six selected genes.
gene_list <- c("CNP","EIF5B","HOPX","KRT19","PHB","SHISA5")
global_results <- spacelink(
normalized_counts = normalized_counts[gene_list,],
spatial_coords = spatial_coords
)
print(global_results[order(global_results$ESV_adj, decreasing=T),c("time","pval","padj","ESV","ESV_adj")])
## time pval padj ESV ESV_adj
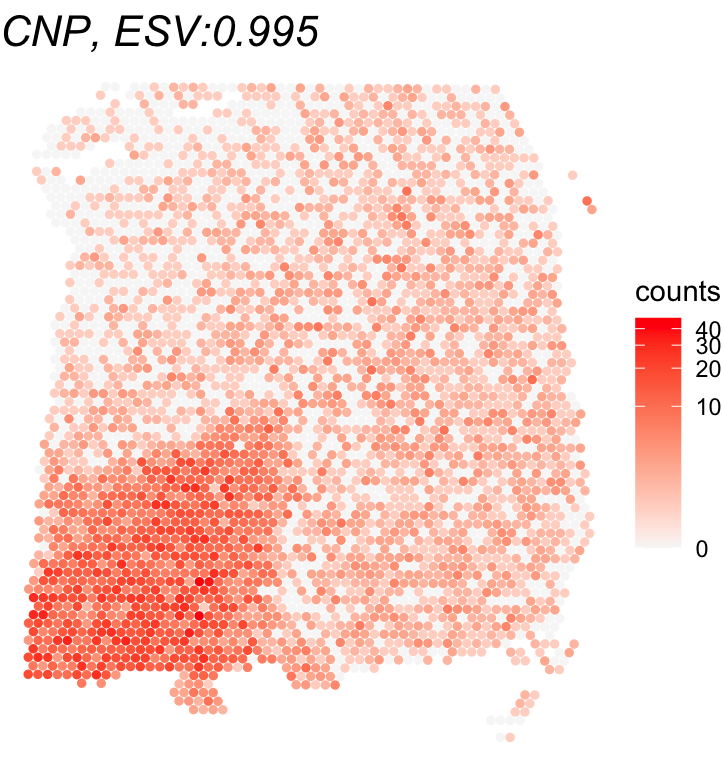
## CNP 4.082 0.00000000 0.000000000 0.982947579 0.982947579
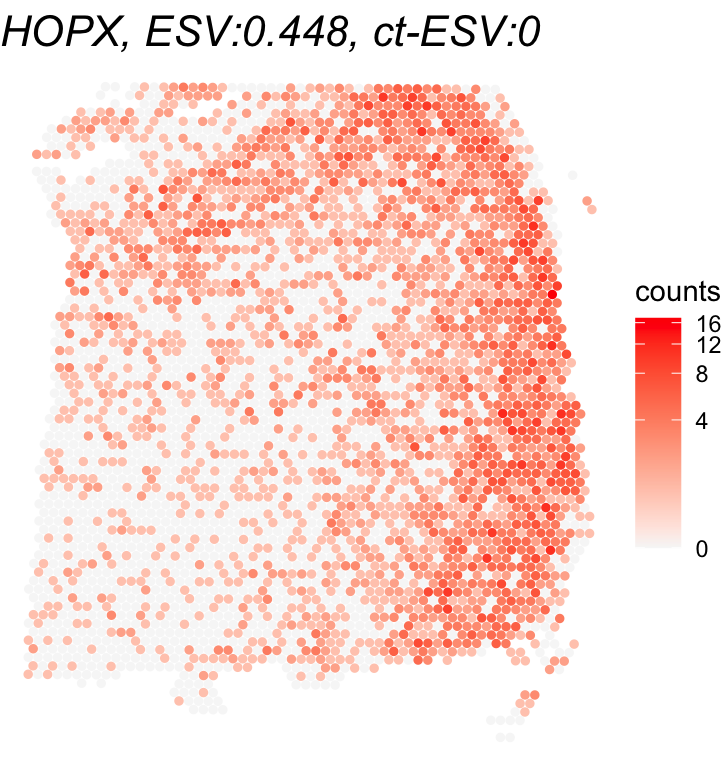
## HOPX 3.904 0.00000000 0.000000000 0.709631417 0.709631417
## KRT19 3.792 0.00000000 0.000000000 0.684495108 0.684495108
## EIF5B 3.736 0.00373013 0.005595195 0.025783391 0.025783391
## SHISA5 3.736 0.02000210 0.024002518 0.009663969 0.009663969
## PHB 3.734 0.10824339 0.108243395 0.026795125 0.0000000005. Visualize Global Spacelink Results
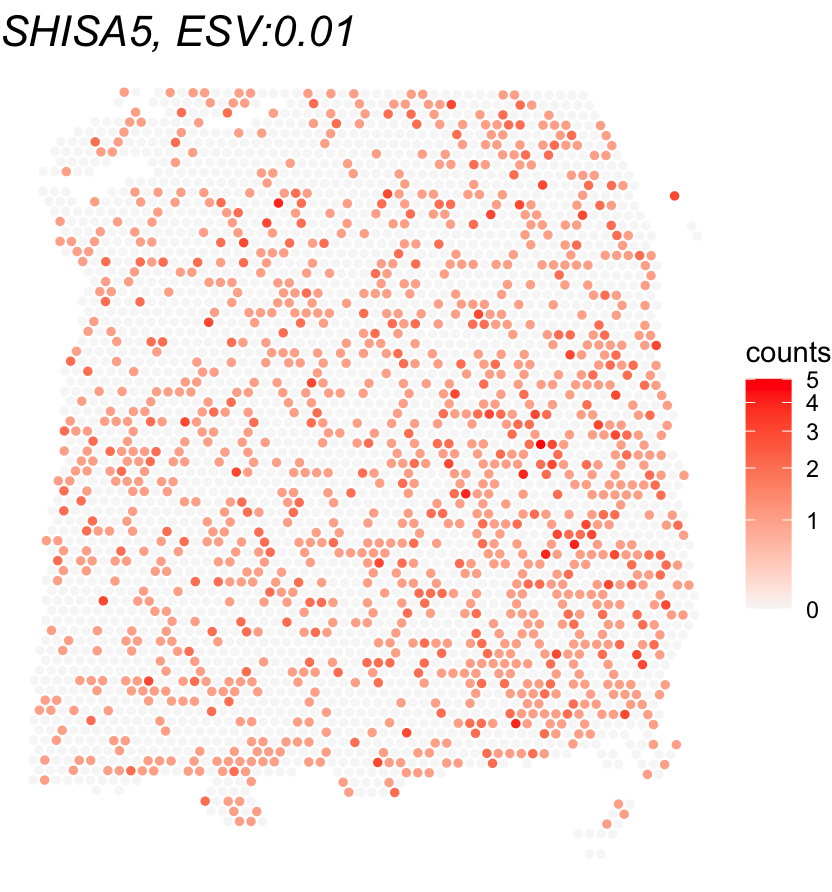
To get an intuitive sense of what the ESV score means, let’s plot the gene with the highest ESV alongside the one with the lowest. This gives us a side-by-side comparison of a strong SVG vs. a non-SVG, showing how differently their expression is distributed across the tissue.
gene_names <- rownames(global_results)[c(which.max(global_results$ESV_adj),
which.min(global_results$ESV))]
for(gene_name in gene_names){
df <- as.data.frame(cbind(spatial_coords, expr = counts[gene_name,]))
colnames(df) <- c('X0', 'X1', 'expr')
title <- paste0(gene_name, ", ESV:", round(global_results[gene_name,'ESV'],3))
print(ggplot(df, aes(x = X0, y = X1, color = expr)) +
geom_point(size = 1) +
ggtitle(title) + theme_void() + scale_y_reverse() +
scale_color_gradient(low = "grey97", high = "red", trans = "log1p", name = "counts") +
theme(plot.title = element_text(face = "italic", size = 16)))
}
6. Run Cell-type-specific Spacelink
Here, we dig a little deeper by asking whether a gene’s spatial
pattern emerges within a particular cell type. We use spacelink_ctSVG
with oligodendrocyte as our focal cell type. The ct-ESV score
in the output works the same way as ESV, but specifically reflects
variability tied to that cell type. Again, note that the
padj and ESV_adj values will differ when
running the analysis on the full gene set, because the
Benjamini–Hochberg adjustment is applied across all genes. In this
example, the adjustment is performed using only the six selected
genes.
cell_type_results <- spacelink_ctSVG(
normalized_counts = normalized_counts[gene_list,],
spatial_coords = spatial_coords,
cell_type_proportions = cell_type_proportions,
focal_cell_type = "oligodendrocyte"
)
print(cell_type_results[order(cell_type_results$ESV_adj, decreasing=T),])
## time pval padj ESV ESV_adj
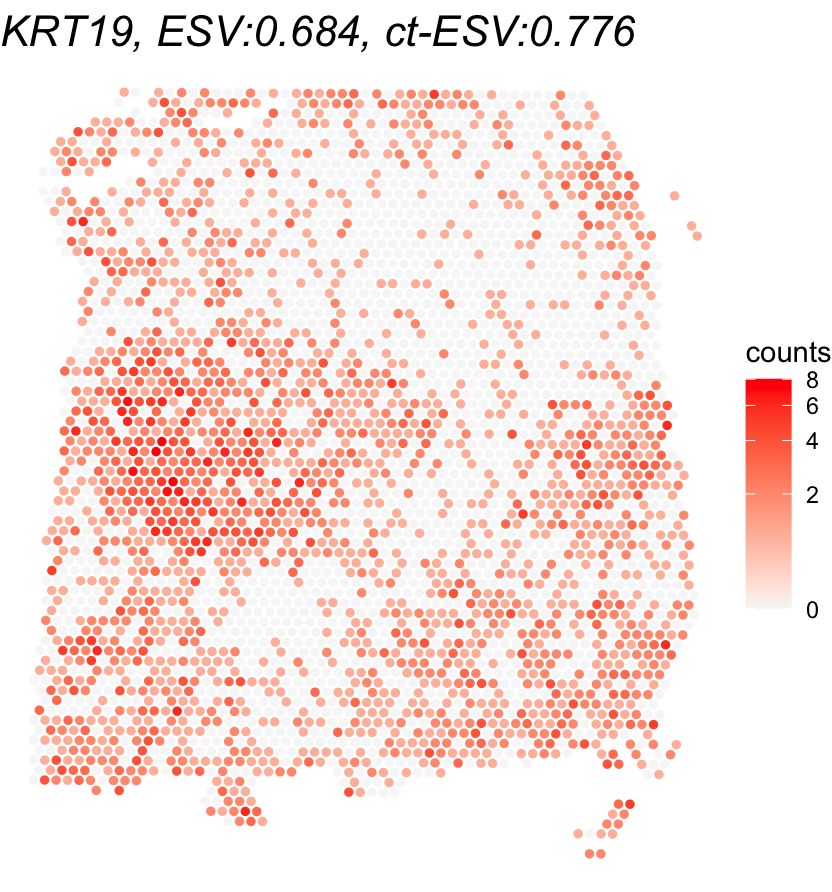
## KRT19 112.470 2.295822e-06 6.887466e-06 7.762241e-01 0.7762241
## CNP 130.494 7.442970e-07 4.465782e-06 6.518893e-01 0.6518893
## EIF5B 148.584 2.066347e-01 4.132695e-01 2.195354e-01 0.0000000
## HOPX 121.434 9.434479e-01 9.434479e-01 2.236363e-05 0.0000000
## PHB 144.309 3.258193e-01 4.887289e-01 4.234864e-02 0.0000000
## SHISA5 149.888 6.209340e-01 7.451208e-01 1.455330e-02 0.00000007. Visualize Cell-type-specific Spacelink Results
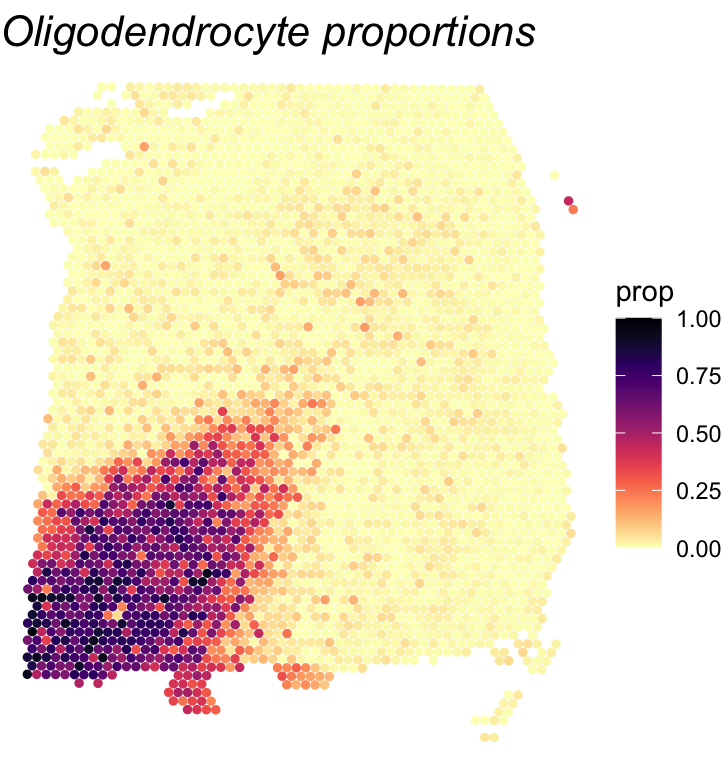
To wrap up, we visualize the results of the cell-type-specific analysis. We first plot the oligodendrocyte proportions across the tissue to see where this cell type is concentrated. Then, we compare the gene with the highest ct-ESV to the one with the lowest. Looking at both ESV and ct-ESV together helps us understand whether a gene’s spatial pattern is really being driven by oligodendrocytes or by something else entirely.
gene_names <- rownames(cell_type_results)[c(which.max(cell_type_results$ESV_adj),
which.min(cell_type_results$ESV))]
# Visualize oligodendrocyte proportions
df <- as.data.frame(cbind(spatial_coords, expr = cell_type_proportions[,'oligodendrocyte']))
colnames(df) <- c('X0', 'X1', 'prop')
print(ggplot(df, aes(x = X0, y = X1, color = prop)) +
geom_point(size = 1) +
ggtitle("Oligodendrocyte proportions") + theme_void() + scale_y_reverse() +
scale_color_viridis(option="magma", direction=-1, limits=c(0,1)) +
theme(plot.title = element_text(face = "italic", size = 16)))
# Visualize gene expression (compare ESV vs ct-ESV)
for(gene_name in gene_names){
df <- as.data.frame(cbind(spatial_coords, expr = counts[gene_name,]))
colnames(df) <- c('X0', 'X1', 'expr')
title <- paste0(gene_name,
", ESV:", round(global_results[gene_name,'ESV'],3),
", ct-ESV:", round(cell_type_results[gene_name,'ESV'],3))
print(ggplot(df, aes(x = X0, y = X1, color = expr)) +
geom_point(size = 1) +
ggtitle(title) + theme_void() + scale_y_reverse() +
scale_color_gradient(low = "grey97", high = "red", trans = "log1p", name = "counts") +
theme(plot.title = element_text(face = "italic", size = 16)))
}